J. Feld (Canada) and H.L.A. Janssen (Canada/Netherlands)
Z. Abbas (Pakistan)
A. Elewaut (Belgium)
P. Ferenci (Austria)
V. Isakov (Russia)
A.G. Khan (Pakistan)
S.G. Lim (Singapore)
S. Locarnini (Australia)
S.K. Ono (Brazil)
J. Sollano (Phillippines)
C.W. Spearman (South Africa)
C.T. Yeh (Taiwan)
M.F. Yuen (Hong Kong)
A.W. LeMair (Netherlands)
1. Introduction
The hepatitis B virus (HBV) causes acute and chronic liver disease and is endemic in many areas of the world. The virus is transmitted through contact with blood or other body fluids from an infected person.
- When transmission occurs vertically (from mother to child) or horizontally to small children (during play, from household contacts etc), the infection usually becomes chronic.
- By contrast, when transmission occurs in adolescents/adults—usually via sexual contact, contaminated needles (“sharps”), and less often from transfusion of blood products—the infection usually resolves unless the individual is immunocompromised (e.g., infected with human immunodeficiency virus).
- Education about how to avoid risky behavior plays an important role in HBV prevention.
- HBV is an important occupational hazard for health-care workers.
- A safe and effective vaccine for HBV has been available since 1982 and is 95% effective at preventing new infections.
Every individual with chronic HBV infection (CHB) represents an opportunity for further cases to be prevented. It is important to take the time needed to educate patients and to explain the risks that the infection poses to the patients themselves and to others.
- HBV vaccination is highly effective, and universal vaccination at a young age—preferably at birth in high-endemicity countries—is desirable.
- At the very least, vaccination should be offered to all individuals who are at risk.
- Pregnant women must be screened for HBV before delivery, as this offers an opportunity to prevent another generation of chronically infected persons.
Although most patients with CHB do not develop hepatic complications, all infected individuals are at increased risk of progressive liver fibrosis, leading to cirrhosis and ultimately to hepatic decompensation and/or hepatocellular carcinoma (HCC). Fortunately, effective treatment can reduce the risk of HBV-related complications.
1.1 WGO Cascades
This Global WGO Guideline includes a set of cascades to provide resource-sensitive options for the diagnosis and management of hepatitis B. These WGO Cascades are intended to serve as a “global” complement to, rather than a replacement for, the “gold standard” guidelines from the European Association for the Study of the Liver (EASL), the American Association for the Study of Liver Diseases (AASLD), the Asian–Pacific Association for the Study of the Liver (APASL), and the National Institute for Care and Health Excellence (NICE) [1–4].
1.2 Epidemiology and transmission of hepatitis B
Of the many viral causes of human disease, few are of greater global importance than hepatitis B virus [5]:
- More than 2 billion people alive today have serologic evidence of past or present HBV infection.
- 250 million are chronically infected and are at risk of developing HBV-related liver disease [6].
- Some 15–40% of chronically infected patients will develop cirrhosis, progressing to liver failure and/or HCC during their lifetime.
- Every year, there are over 4 million acute clinical cases of HBV.
- An estimated 1 million people die each year from chronic HBV infection and its complications: cirrhosis or primary liver cancer [7].
- HBV-related liver deaths (2010) are estimated at 786,000 annually [8].
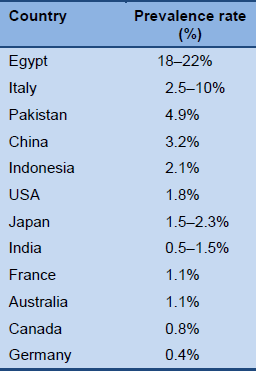
The prevalence of HBV varies markedly between different regions of the world (Fig. 1). In the literature, a distinction is usually made between areas of high, medium, low, and very low endemicity.
- In high-endemicity areas [5], approximately 70–90% of the population become infected with HBV before the age of 40, and 8–20% of people develop chronic infection with persistent carriage of the virus [9].
- The prevalence of CHB ranges from over 10% of the population in South-East Asia, China, the Amazon area, and sub-Saharan Africa to less than 1% in Western Europe and North America.
- Overall, approximately 45% of the global population live in areas of high endemicity. With globalization, many individuals with HBV are immigrating to areas in which the CHB rate has traditionally been low and the condition may easily go unnoticed.
Fig. 1 Geographic distribution of hepatitis B virus genotypes worldwide (reproduced with permission [10,11]). With immigration patterns, the HBV genotype distribution for chronic hepatitis B may change rapidly, particularly in the Western world.
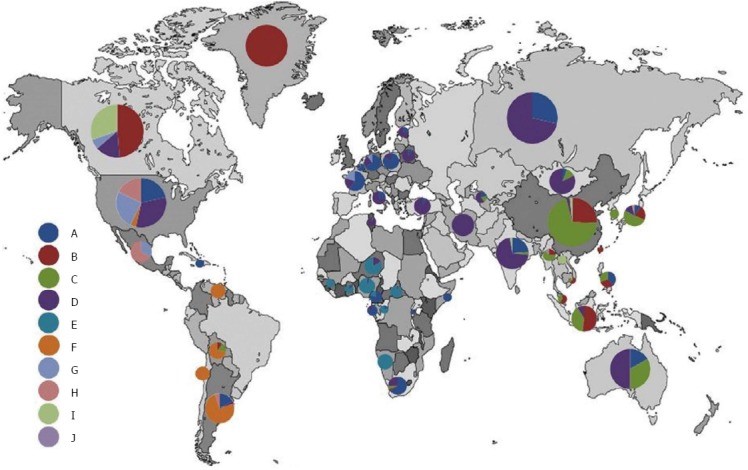
Notes: Recently published data [12,13] demonstrated the following genotype distribution for Russia: genotype D, 85%; genotype A, 10.7%; genotype C, 3.2%; all other genotypes, 1.1%. In Venezuela, HBV genotype F is the most frequent one in the general population (as in Colombia and Peru) [14]; the prevalence in urban populations is approximately 80% [15], while in “Amerindians” it is almost 100% [16].
The wide range of prevalence figures for CHB infection is largely related to differences in age at infection.
- The chance that acute infection will become chronic is 70–90% for perinatally acquired (vertical) infection and 20–50% for (horizontal) infections acquired during early childhood (under the age of 5 years).
- The chance of developing CHB is in the range of 1–3% in immunocompetent adult-acquired HBV infections, with higher rates in immunosuppressed individuals.
- Eight [17] (and possibly up to 10) genotypes of hepatitis B virus have been identified (A–H), which differ in their geographic distribution and in their potential to affect the clinical course of disease (Table 1). The current prevalence of HBV genotypes in different regions is highly dependent on immigration patterns.
Table 1 Geographical distribution of genotypes and subgenotypes of hepatitis B infection [10]
Increasing numbers of patients with chronic infection are developing HBV variants that express little or no hepatitis B e antigen (HBeAg); this HBeAg-negative form of hepatitis B may require long-term therapy to reduce the likelihood that liver disease will progress, with relapse occurring when the patient is off treatment. A distinction is made between a precore mutation, in which a stop mutation in the precore gene completely eliminates HBeAg production, and the basal core promoter (BCP) mutation, which affects the promoter and thus reduces, but does not eliminate, HBeAg production. The prevalence of precore mutations is highest in the Mediterranean countries and they are most prevalent in genotype D, while the core promoter mutations are mostly found in genotype C (in East Asia and South-East Asia).
2. Clinical Course of HBV Infection
The outcome of HBV infection largely depends on the host–virus interaction, mediated by the adaptive immune response. The virus-specific T cell response is one of the key factors in the pathogenesis of HBV infection. Viral variants may influence the course and outcome of the disease. The effect of host factors on the progression of disease is poorly understood. Only very rarely (when there is profound immune suppression) does the hepatitis B virus probably become directly cytopathic.
2.1 Natural history
The clinical course of HBV infection is variable and includes acute (self-limiting) infection, fulminant hepatic failure, inactive carrier state, and chronic hepatitis with chances of progression to cirrhosis and HCC [24,25].
2.2 Chronic HBV infection
The risk of chronicity in acute HBV infection is related to age at primary infection. Adults who become chronically infected during childhood have a 15–25% lifetime risk of dying from HBV-related cirrhosis or liver cancer, with a significantly increased risk in men in comparison with women [26].
Table 2 Risk of chronicity and age at primary infection

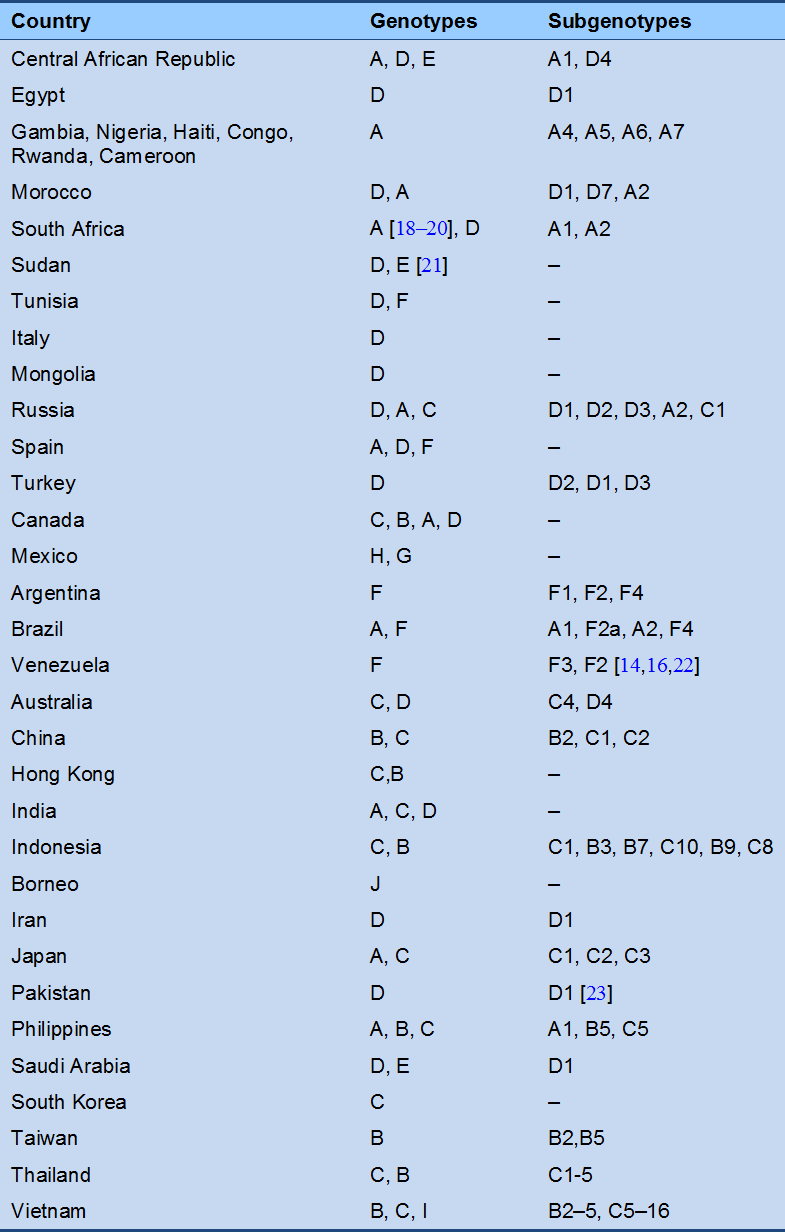
Fig. 2 Sequence of serologic markers in acute hepatitis B infection (reproduced with permission from [27]). A, resolution of active infection; B, progression to chronic infection.
2.3 CHB disease phases
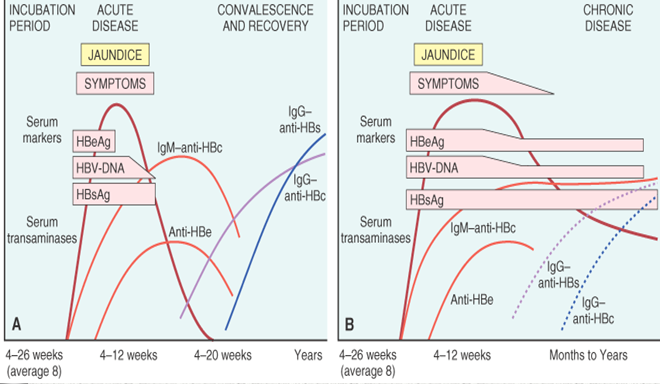
CHB is a dynamic disease that fluctuates over time, likely relating to interactions between the virus and the host immune system. The following five—not necessarily sequential—phases can be identified in chronic HBV infection.
- Immune-tolerant phase:
— Characterized by high levels of serum HBV DNA, HBeAg positivity, normal alanine aminotransferase (ALT) levels, and absent liver necroinflammation.
— Disease progression is minimal in patients who remain in this phase [28].
— Patients are highly contagious in this phase.
- Immune-reactive phase (HBeAg-positive CHB):
— Patients enter this phase after a variable time, linked to the age when HBV infection occurred.
— The immune system becomes more active and the infected hepatocytes are attacked.
— Characterized by highly fluctuating, but progressively decreasing, HBV-DNA levels, elevated ALT, and hepatic necroinflammation (HBeAg-positive CHB).
— A prolonged immune-active phase with multiple ALT flares may result in progressive liver fibrosis, leading to cirrhosis.
- Immune-control phase (and inactive carrier state):
— Transition into this phase as an outcome of the immune-active phase is marked by seroconversion from HBeAg to anti-HBe positivity.
— Characterized by low (< 2000 IU/mL) or undetectable serum HBV DNA, normal ALT levels, and disappearance of liver necroinflammation (inactive carrier state).
- Reactivation phase (HBeAg-negative CHB):
— Despite HBe seroconversion, reactivation of HBV replication may occur due to the selection of HBeAg-defective HBV mutants.
— Characterized by positive anti-HBe antibody levels, fluctuating HBV DNA and ALT levels, and a high risk of progression to severe hepatic fibrosis (HBeAg-negative CHB).
— Periodic ALT flares with intervening normalization may make it difficult to distinguish between HBeAg-negative CHB and inactive disease, and thus continued follow-up is required before patients with normal ALT and low HBV DNA levels are designated as inactive carriers.
— Emerging evidence suggests that a low HBV DNA titer (< 2000 IU/mL) combined with a low hepatitis B surface antigen (HBsAg) titer (< 1000 IU/mL) may help identify inactive carriers, particularly those with genotype D infection [29].
- HBsAg-negative phase:
— After HBsAg loss, low-level HBV replication may persist, with detectable HBV DNA in the liver and rarely in the serum [30].
— In patients with “occult” HBV infection, persistence of effective HBV immunological control has been demonstrated [31].
— Significant immunosuppression may lead to HBV reactivation, with reappearance of HBsAg, known as “reverse seroconversion.”
Fig. 3 Markers and natural history of chronic hepatitis B infection (reproduced with permission from [27]). 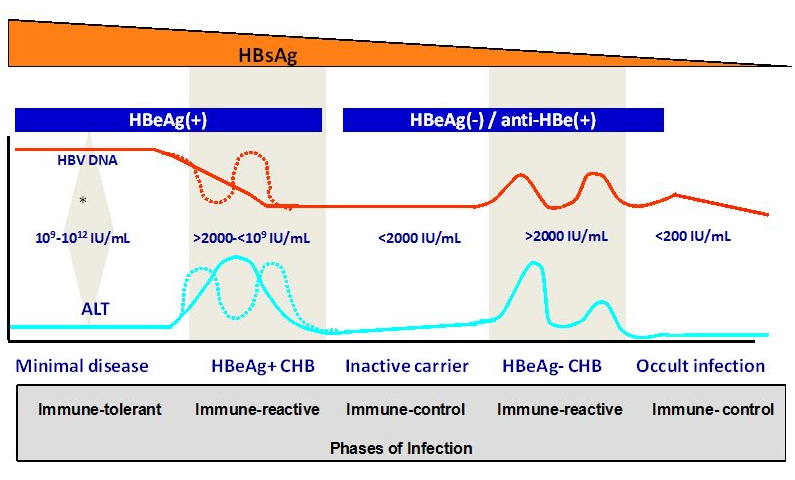
2.4 Progression of CHB
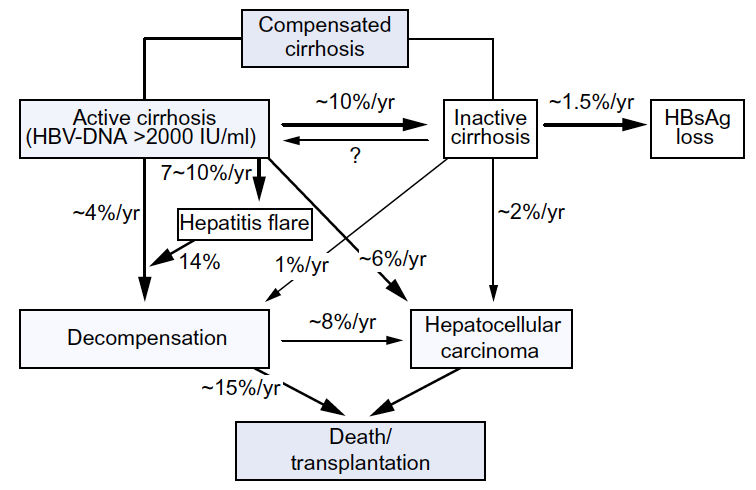
CHB has a very variable course, ranging from silent subclinical infection to persistent hepatitis with progressive fibrosis leading to cirrhosis, liver failure and/or liver cancer. The determinants of disease outcome are incompletely understood, but include viral, host, and environmental factors (Table 3), all of which interact. Viral determinants of the prognosis have different significance depending on the stage of the disease. For example, serum HBV DNA titers are highest in the immune-tolerant phase of disease, despite the lack of hepatic inflammation or progressive fibrosis during this period. In contrast, in HBeAg-negative CHB, the higher the HBV DNA level, the greater the risk of disease progression and HCC. The rates of progression to cirrhosis and HCC and associated mortality rates are shown in Fig. 4.
Table 3 Factors in the disease outcome with chronic hepatitis B
BCP, basal core promoter; HBeAg, hepatitis B e antigen; HBV, hepatitis B virus; HCC, hepatocellular carcinoma; HCV, hepatitis C virus; HDV, hepatitis D virus; HIV, human immunodeficiency virus.
Fig. 4 Risk of progression in patients with HBV-related cirrhosis. Reproduced with permission from Peng et al. (2012) [32], to which reference may be made for a detailed discussion of the natural course of HBV-related cirrhosis and HCC.
3. Diagnosis and monitoring of hepatitis B
3.1 Cascade - acute hepatitis B
The diagnosis of acute hepatitis B is based on the detection of HBsAg and anti-HBc (immunoglobulin M).
- During the initial phase of infection, markers of HBV replication—HBeAg and HBV DNA—are also present.
- Recovery is accompanied by the disappearance of detectable HBV DNA, HBeAg seroconversion to anti-HBe, and subsequent clearance of HBsAg with seroconversion to anti-HBs and appearance of anti-HBc (IgG).
- The course of acute HBV should take place within 3 months of the diagnosis—chronic HBV infection is characterized by persistence of plasma HBsAg for more than 6 months.
Rarely, patients present during the window period when HBsAg has already become negative but anti-HBs is not yet positive. In this setting, which is more common in patients with fulminant hepatitis B, in whom viral clearance tends to be more rapid, immunoglobulin M (IgM) anti-HBc is the sole marker of acute HBV infection.
Cascade 1 Diagnostic tests for acute hepatitis B

The differential diagnosis of HBsAg-positive acute hepatitis includes exacerbations of CHB, which may occur at any time in any individual who is chronically infected (at these times, reversion back to anti-HBc IgM may occur). Acute hepatitis may occur following withdrawal from immunosuppressive therapy or through superinfection of a person chronically infected with hepatitis B with hepatitis C and/or D virus, or hepatitis A virus. Superimposed acute hepatitis due to drugs and other toxins administered to someone who has “silent” CHB infection may also present as acute hepatitis. A precipitating factor is sometimes not identified.
3.2 Resolved HBV infection
Previous HBV infection is characterized by the presence of anti-HBs and IgG anti-HBc. Anti-HBs sometimes becomes undetectable after many years. (Anti-HBs is frequently undetectable if HBV infection occurred during childhood, as is seen in sub-Saharan Africa). Notably, although these individuals are referred to as having “resolved HBV” infection, trace amounts of HBV DNA remain in their livers for years and possibly even lifelong. Immune control prevents viral expansion, but means that with severe immunosuppression (e.g., with advanced human immunodeficiency virus (HIV) coinfection, bone marrow transplantation, rituximab use, etc.), HBsAg may reappear (reverse seroconversion) or viral replication may be detectable in the liver even without the reappearance of serum HBV DNA. Immunity to HBV infection after vaccination is characterized by the presence of only anti-HBs.
3.3 Chronic HBV infection
Diagnosis of chronic HBV infection is defined as the persistence of HBsAg for more than 6 months.
- It must first be established whether the individual is in the HBeAg-positive or HBeAg-negative phase of the infection (Table 4).
- Additional tests for markers of HBV replication—namely, HBeAg and serial measurements of serum HBV DNA, in addition to ALT—should be carried out.
- This will in part determine whether the patient should be considered for HBV therapy.
- Both HBeAg-positive and HBeAg-negative patients, even if they have normal serum ALT (women < 20 IU/L and men < 30 IU/L) and/or undetectable HBV DNA, still need to be monitored lifelong, as the condition may change over time even if they remain asymptomatic.
- Among individuals with chronic persistence of HBsAg, those with elevated serum ALT concentrations should be followed more closely, preferably with serial HBV DNA measurements.
- It is important to know the lower limit of detection of the method used to measure HBV DNA, as values that are persistently ≥ 2000 IU/mL will prompt consideration of antiviral therapy.
- The decision on whether to initiate therapy depends on multiple factors (i.e., not just the level of HBV DNA and/or ALT). If the liver disease appears to be progressing (as judged by liver biopsy and noninvasive markers of inflammation and fibrosis such as transient elastography), treatment should be considered.
- Additional tests for hepatitis C and hepatitis D should also be conducted in order to rule out superinfection with other hepatitis virus(es), particularly in patients with elevated ALT but low or undetectable HBV DNA.
- Other things to consider include drug-induced liver injury (due to supplements), nonalcoholic steatohepatitis (NASH), and iron overload.
Table 4 Differentiation of phases of CHB infection
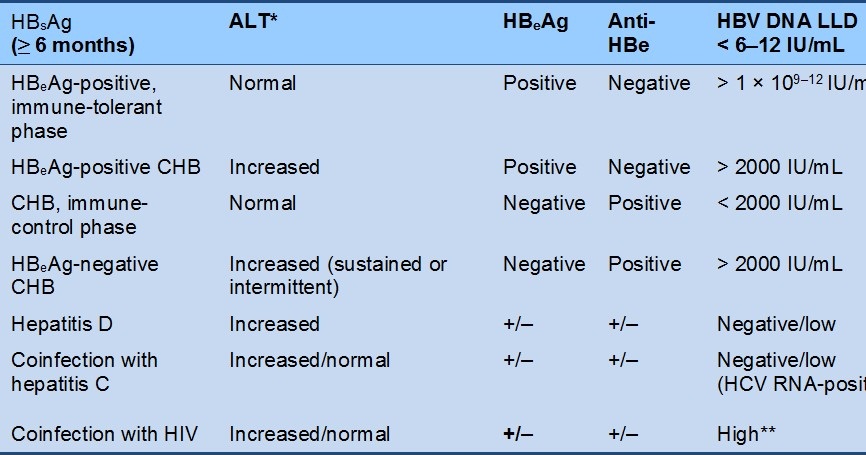
CHB, chronic hepatitis B; HBeAg, hepatitis B e antigen; HBsAg, hepatitis B surface antigen; HBV, hepatitis B virus; HIV, human immunodeficiency virus; LLD, lower limit of detection.
*Normal range < 20 IU/L in women, < 30 IU/L in men.
** May be variable, depending on the mode and age of acquisition of HBV, HIV/HBV coinfection, and on the CD4 count.
Different levels of HBV DNA are used for initiating treatment for HBeAg-positive and HBeAg-negative disease, depending on the genotype prevalent in different regions. As a general rule (and because genotyping all patients is not feasible), the EASL level can be used for Caucasian patients: 2 × 103 IU/mL level (and age > 30 y); and the APASL/AASLD level can be used for Asian patients: 2 × 104 IU/mL (and age > 40 y).
3.4 Initial evaluation of patients with chronic HBV infection
Individuals with newly detected CHB need to understand that long-term monitoring for the development of chronic hepatitis, cirrhosis, and HCC via a series of clinical examinations and laboratory tests is required even if they are asymptomatic. It is important to verify the stage of CHB and decide the frequency of follow-up examinations needed.
- Chronic HBV infection is not necessarily accompanied by progressive liver disease requiring antiviral therapy.
- Accurate evaluation of all HBsAg-positive carriers is required in order to identify [33]:
— Phase of infection
— Grade of liver inflammation
— Stage of liver fibrosis
— Concurrent causes of liver disease
— Need for treatment
— Presence of cofactors increasing the risk of progression to cirrhosis or HCC: coinfections with hepatitis D virus (HDV), hepatitis C virus (HCV), and HIV; comorbidities including alcoholism, autoimmune disease, or metabolic liver disease
The initial examination should include:
- History and physical examination, including skin and abdominal examination.
- Markers of HBV infection, including: HBeAg/anti-HBe and HBV DNA to classify the phase of CHB, as well as the HBV genotype if antiviral therapy with interferon is contemplated.
- Markers of other viral infections, including HCV and HDV, particularly if ALT is elevated but HBV DNA is low or undetectable.
- Before oral antiviral therapy is introduced, all patients should be screened for human immunodeficiency virus (HIV).
- Complete liver panel (ALT/AST to identify active inflammation, and bilirubin, prothrombin time, and albumin to check liver synthetic function).
- Complete blood count, particularly platelets, which serve as a surrogate marker for portal hypertension.
- Abdominal ultrasonography for baseline screening for HCC—alpha fetoprotein may be used in areas with high HBV endemicity and poorly differentiated HCC, as well as in areas without easy access to high-quality ultrasound.
- Measurement of liver fibrosis by serological testing, FibroScan (transient elastography), or liver biopsy.
Table 5 Host and viral risk factors associated with progression of chronic hepatitis B

ALT, alanine aminotransferase; HBeAg hepatitis B e antigen; HBV, hepatitis B virus; HCC, hepatocellular carcinoma; HDV, hepatitis D virus; HIV, human immunodeficiency virus; NAFLD, nonalcoholic fatty liver disease.
3.5 Occult HBV
Occult HBV infection refers to the persistence of HBV DNA in liver tissue (and in some cases in serum) in individuals in whom hepatitis B surface antigen (HBsAg) is not detectable in the blood, usually with positive anti-HBc.
Occult HBV infection is prevalent worldwide, but its frequency is related to the prevalence of overt HBV infection in a specific geographic area. Occult HBV is transmissible through blood transfusions and organ transplantation.
- Blood products should be screened for HBsAg, anti-HBc, and ideally HBV DNA.
- Organs from donors with anti-HBc and/or anti-HBs should preferably be used only for recipients who test positive for anti-HBs or HBsAg.
Although the true relevance of occult HBV infection is unknown, it may be an additional risk factor for HCC in anti-HCV–positive patients and in HIV-infected individuals. It may also be associated with progression of chronic liver disease due to causes other than HBV.
3.6 HBV reactivation
HBV replication is controlled by the host immune system. Immune suppression of any kind can lead to a loss of immune control and subsequent HBV reactivation, which can result in a range of consequences, from a subclinical increase in HBV DNA to icteric and even fulminant and/or fatal liver failure. Reactivation occurs most frequently with cancer chemotherapy, but may occur with other immunosuppressive or immunomodulator therapy (e.g., targeted immunotherapy). The addition of systemic corticosteroids (SCS) to inhaled corticosteroids increases the risk of HBV reactivation, especially when SCS are administered chronically or at high doses [35].
Preemptive treatment with a nucleoside/nucleotide analogue is recommended in HBsAg-positive patients who are going to receive anticancer or immunosuppressive drugs. Treatment should continue throughout the course of immunosuppression and for 6–12 months afterwards, with follow-up monitoring to ensure that flares do not occur upon withdrawal of antiviral therapy.
Reactivation may also occur in patients who are HBsAg-negative but anti-HBc-positive (with or without occult HBV DNA), but more significant immune suppression is required. Reappearance of HBsAg is referred to as reverse seroconversion. The risk appears to be increased with rituximab or other anti-CD20–based chemotherapy, probably due to the long-lasting depletion of B cells. HBV DNA may increase even before HBsAg reappears in the serum. Preemptive treatment with a nucleoside/nucleotide analogue reduces the risk of HBV reactivation, but may not be required in all patients [36]. Those who are not preemptively treated should be monitored with serial HBsAg, ALT, and possibly HBV DNA with antiviral therapy being started if HBsAg reappears or HBV DNA increases.
In summary:
- Screening for HBsAg and anti-HBc is necessary before chemotherapy or immunosuppressive/immunomodulator therapy is started.
- Patients who are HBsAg-positive should receive preemptive antiviral therapy during and for 6–12 months after chemotherapy.
- The benefits of preemptive treatment for occult HBV reactivation remain unclear at present.
- For patients with evidence of previous HBV infection, as confirmed by positive anti-HBc with or without anti-HBs, serial monitoring of HBV-related markers is recommended during and after immunosuppressive therapy.
- Patients receiving chemotherapy or immunosuppression should follow the American Association for the Study of Liver Diseases (AASLD) and Asian-Pacific Association for the Study of the Liver (APASL) guidelines (Fig. 5).
Fig. 5 APASL algorithm for all candidates for chemotherapy.

NA, nucleoside analogue.
Source: Asian–Pacific Association for the Study of the Liver.
3.7 HCC screening
The aim is to detect tumors smaller than 3 cm in diameter, and preferably less than 2 cm, in order to offer a potential for curative treatment. Screening for HCC is advocated in all cirrhotic patients, as they are at the highest risk of developing HCC. However, in Africa and South-East Asia, where HBV infection is acquired early in life, HCC may develop in a noncirrhotic liver.
The AASLD recommends HCC surveillance in the following types of patients with CHB:
- Asian men over the age of 40 and Asian women over the age of 50
- All patients with cirrhosis, regardless of age
- Patients with a family history of HCC; any age
- Africans over the age of 20
- Any individuals with HBV/HIV coinfection
Singal et al. showed that in a “real-world” clinical setting, a combination of ultrasound and alpha fetoprotein (AFP) is the most effective strategy for detecting HCC at an early stage. The sensitivity significantly improved to 90%, with a minimal loss of specificity (83%). AFP alone may be better than ultrasound alone, as the reliability of ultrasound is very dependent on the skill and experience of the ultrasonographer [37].
For hepatitis B carriers not included in this list, the risk of HCC varies depending on the severity of the underlying liver disease and current and past hepatic inflammatory activity. Those with high HBV DNA concentrations and ongoing hepatic inflammatory activity (evidenced by elevated ALT values) are at increased risk for HCC, and surveillance should be considered. Genotype C infection and the presence of BCP and pre-S1 mutations are also associated with an increased risk of HCC.
4. Treatment for CHB
Before any form of HBV therapy is started, and optimally at the time of first presentation, the patient needs to be provided with information about CHB and its treatment. Important information includes:
- The dynamic clinical course of CHB.
- Most infections remain initially entirely asymptomatic, even in the case of severe disease.
- The need for regular lifelong monitoring.
- Possible transmission to contacts—family and contacts need HBV screening and vaccination of those who are not immune to HBV, and referral for clinical evaluation of those who are HBsAg-positive.
- Timing of the start of treatment.
- The need for absolute compliance with potentially long-term therapy.
- The need for absolute compliance with follow-up examinations both when the patient is on treatment and when he/she is off treatment.
- The importance of alcohol abstinence and attention to the use of medications that may be hepatotoxic or dangerous in patients with advanced liver disease (e.g., NSAIDs) should be emphasized.
- Those who are not immune to hepatitis A should receive two doses of hepatitis A vaccine 6–18 months apart.
This information should be explained and discussed with the patient. In women of childbearing potential, drugs that are considered safe in pregnancy are preferred, because once a nucleoside or nucleotide has been prescribed it cannot be stopped in those who remain HBeAg-positive. The patient needs to understand that cessation of treatment may precipitate severe hepatitis, which can, rarely, lead to fulminant acute liver failure, even in the absence of cirrhosis.
The phase of CHB can be determined on the basis of the serological and virological profile—each type is characterized by a distinct natural course, prognosis, and treatment indications [1,2,38]
1 Immune-tolerant carrier:
- Treatment not indicated.
- Appropriate longitudinal follow-up is crucial.
- Measure ALT every 3–6 months.
2 Inactive carrier:
- Treatment not indicated.
- Appropriate longitudinal follow-up is crucial.
- Assess ALT and HBV DNA levels every 3 months during the first year, then every 6 months.
- If the serum HBV DNA is < 2000 IU/mL and the HBsAg level is < 1000 IU/mL, the probability of disease reactivation is low and patients may require less frequent monitoring.
3 Active CHB:
- HBeAg-positive CHB.
- HBeAg-negative CHB..
The prognosis and management of CHB greatly depend on the phase of the disease and the stage of liver fibrosis, and thus the risk of cirrhosis developing. Follow-up in CHB HBsAg carriers includes:
- Continuation of diagnostic work-up.
- Assessment of the severity of the liver disease.
— Laboratory tests for inflammation (ALT), hepatic function (bilirubin, albumin, coagulation factors) and viral load (HBV DNA), if available.
— Hepatic ultrasound examination.
— Noninvasive measures to assess fibrosis (serum panels, transient elastography).
— Liver biopsy, useful for determining the grade of necroinflammation and the stage of fibrosis.
— Liver biopsy. This can help exclude other coexistent causes of liver disease and clarify the diagnosis when ALT and HBV DNA levels are discordant.
The current standards for deciding on treatment for CHB are shown in Figures 6 and 7. Cascades are included to reflect resource-sensitive options.
Fig. 6 Management of chronic HBeAg-positive infection. Surveillance for hepatocellular carcinoma should be carried out if indicated (depending on age, sex, severity of liver disease, and family history). Adapted from Lok and McMahon 2007 [39].
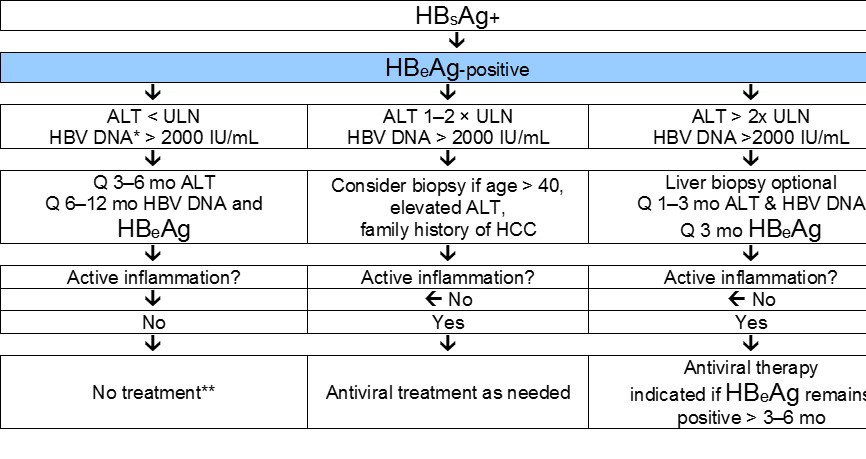
* In patients of Asian origin, very high HBV DNA levels are frequently observed—mostly in patients with perinatal transmission. It is unclear whether they should receive antiviral treatment with NAs. The level of HBV DNA correlates with the risk of HCC during follow-up, but whether viral suppression reduces the risk is unclear.
** Patients with cirrhosis and detectable HBV DNA should be treated regardless of their ALT value and level of HBV DNA.
Fig. 7 Management of chronic HBeAg-negative infection.
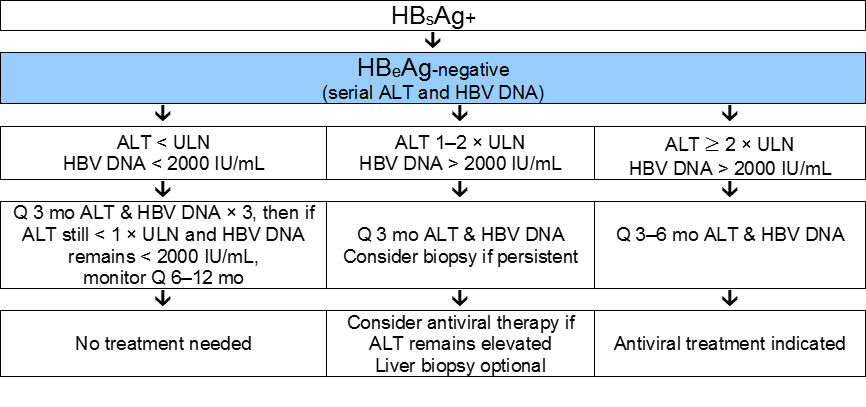
NB: Surveillance for HCC should be carried out if indicated (depending on age, sex, severity of liver disease, and family history).
The upper limit of normal for ALT is 20 IU/L in women and 30 IU/L in men.
Monitoring HBV DNA every 3 months in patients with ALT one to two times the upper limit of normal is expensive and may not be practical when economic resources are limited—see the cascades below for alternative approaches.
4.1 Cascades for CHB management—a resource-sensitive approach
A standard approach is only feasible if the full scale of diagnostic tests and medical treatment options are available. Such resources may not be sufficiently available throughout the world. With their diagnostic and treatment cascades, the World Gastroenterology Organisation guidelines provide a resource-sensitive approach.
- Assessment of the baseline HBV DNA level, HDV and HIV are recommended for all resource levels prior to any therapy.
- Initial HCC assessment using ultrasonography should be done in all cases, where possible. Alpha fetoprotein still has a role in monitoring in resource-poor areas with high HBV endemicity, poorly differentiated HCC, and limited access to high-quality ultrasound.
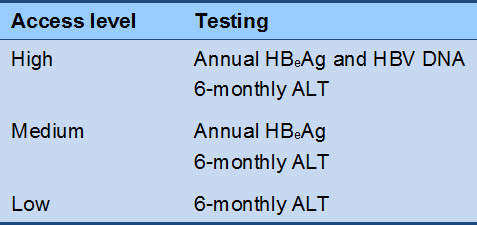
Cascade 2 Immunotolerant phase monitoring (no therapy)
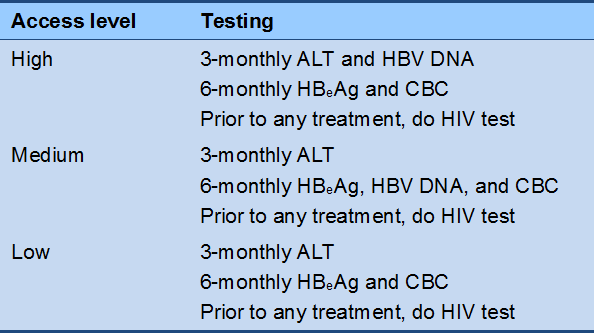
Cascade 3 Immunoactive phase monitoring (off therapy)
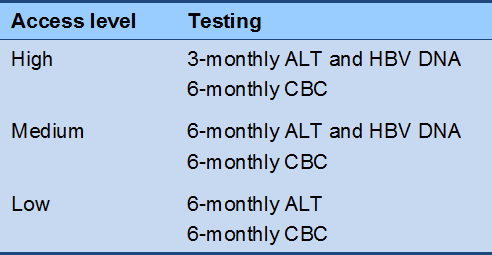
Cascade 4 Immune-control phase monitoring, HBeAg-negative (off therapy)

Cascade 5 Reactivation phase monitoring, HBeAg-negative (off therapy)
Cascade 6 Immunoactive phase monitoring, HBeAg-positive (interferon-based therapy)

* HBsAg titer at week 12—“week 12 interferon stopping rule.”
Cascade 7 Immunoactive phase monitoring, HBeAg-positive (on NA therapy)
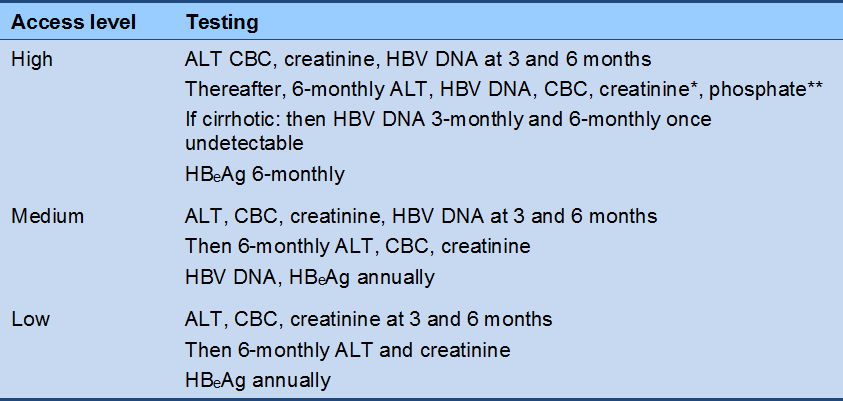
* At all levels: if the patient is receiving tenofovir, the frequency of creatinine testing is guided by renal function.
** Phosphate testing required only for patients receiving tenofovir.
Cascade 8 Reactivation phase monitoring, HBeAg-negative (on NA therapy)

* If the patient is receiving tenofovir, the frequency of creatinine testing is guided by renal function.
** Phosphate testing is required only for patients receiving tenofovir.
4.2 Treatment for CHB
Approved drugs
Table 6 Approved drugs for chronic hepatitis B
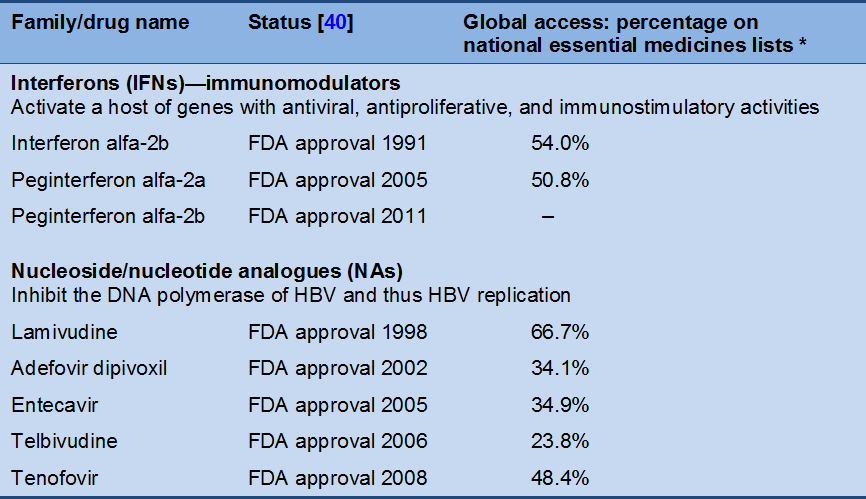
* Reported percentages of WHO member states with drugs for hepatitis B on their national essential medicines lists or subsidized by their governments [41].
Table 7 shows the results of the major studies on the treatment of HBeAg-negative and HBeAg-positive chronic hepatitis B 6 months after completion of 12 months (48 weeks) of pegylated interferon alpha (PEG-IFN) and after 12 months (48 or 52 weeks) of nucleoside/nucleotide analogue therapy.
Table 7 Research results on the treatment of HBeAg-negative and HBeAg-positive chronic hepatitis B
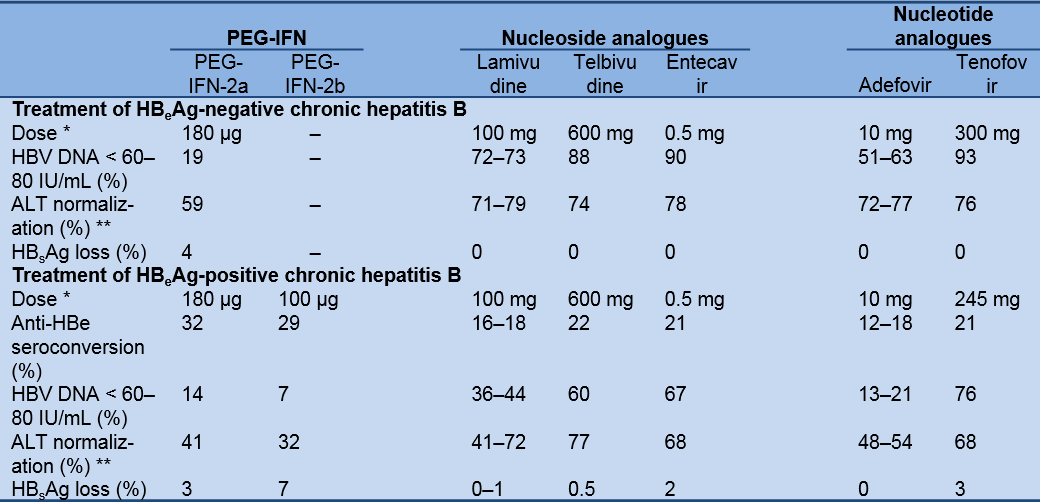
ALT, alanine aminotransferase; HBV, hepatitis B virus; HBsAg, hepatitis B surface antigen; PEG-IFN, peginterferon. Adapted from the 2012 EASL guideline [2]; for further references, the source should be consulted.
* Administration of PEG-IFN: percutaneous injections once weekly; nucleoside/nucleotide analogues: oral tablets once daily.
** The definition of ALT normalization varied among different trials (e.g., with a decrease of ALT up to 1.25 times the upper limit of normal (ULN) in the entecavir trial or up to 1.3 times the ULN in the telbivudine trial).
For a detailed discussion of the “gold standard” treatment for CHB, reference should be to the latest 2012 EASL guideline [2] (www.easl.eu).
Drug resistance
If there is no response or virological breakthrough, as defined by an increase in the HBV DNA level of more than 1 log10 IU/mL in comparison with the nadir (lowest value) HBV DNA level during treatment with confirmed compliance, then another agent with the optimal resistance profile—i.e., tenofovir or entecavir—should be substituted or added.
The following strategies can be used to prevent resistance:
- For the first-line therapy, choose a potent antiviral drug and/or one with a low incidence of resistance (high genetic barrier) over time (entecavir/tenofovir).
- Emphasize to the patient once again the importance of absolute compliance with therapy.
- The HBV DNA level should be monitored frequently when using drugs with a low barrier to resistance (every 3–6 months) during treatment, and resistance testing (genotyping) should be carried out in case of viral breakthrough or suboptimal viral suppression, to allow genotypic resistance to be detected before clinical consequences develop.
- No drug resistance to interferon has been described, although some individuals do not respond to therapy, in which case it should be stopped. If available, the HBsAg titer can be used to guide interferon therapy (see below).
HBeAg-positive hepatitis
Recommendations. HBeAg-positive patients with persistent ALT ≥ 2 × the upper limit of normal, and with HBV DNA ≥ 2000 IU/mL, should be considered for treatment.
- It is imperative to check for HIV coinfection before treatment, because all approved nucleoside/nucleotide analogues have activity against HIV and will rapidly lead to drug-resistant HIV if used as monotherapy.
- HDV testing should be mandatory in countries with a high prevalence of hepatitis D infection (Romania, Moldavia, former Soviet central Asian republics, Russia).
- In patients who have had a liver biopsy, treatment should be started for those with moderate to severe inflammation or significant fibrosis (≥ F2).
- Treatment should be initiated in those who have cirrhosis with detectable HBV DNA, even those with a low HBV DNA level, irrespective of the ALT level.
- Any of the approved therapies can be chosen, and the decision regarding the selection of therapy should include an assessment of efficacy, safety, and genetic barriers to resistance. To avoid resistance, entecavir and tenofovir are the preferred choices for NA therapy. It is important to ensure that patients have a secure source of support to pay for medications over the longer term before starting therapy, to avoid abrupt cessations of treatment, which can be dangerous.
- Patients should be monitored regularly during therapy at 3–6-monthly intervals, or more frequently if they are receiving interferon-based therapy, to monitor for efficacy, safety, and early evidence of resistance (for nucleoside/nucleotide analogues).
- Ideally, patients should be monitored with ALT, HBeAg, anti-HBe, and HBV DNA, but this may not be possible in countries in which these tests are not available or are prohibitively expensive, in which case ALT will have to suffice.
- Virologic breakthrough: an increase in HBV DNA > 1 log above the nadir after a virologic response has been achieved during continued treatment (for nucleoside/nucleotide analogues). Before assuming this is resistance, adherence should be discussed with the patient. A continued increase in the HBV DNA titer over time is suggestive of resistance in a patient who is complying with the treatment.
- Patients with resistance should be considered for rescue therapy with nucleosides/nucleotides that do not have a cross-resistant profile (lamivudine, telbivudine, and entecavir have an overlapping resistance profile, so that tenofovir substitution would be preferable—or if unavailable, adefovir add-on therapy).
- Oral agents should be continued until at least 12 months after the end point of HBeAg seroconversion occurs in HBeAg-positive hepatitis, and it may be preferable to continue until HBsAg loss occurs because of the high risk of reactivation after cessation of therapy. Close monitoring is recommended after oral therapy has been stopped or withdrawn, because of the risk of a treatment withdrawal flare.
- Peginterferon-based therapies have the advantage of a fixed duration of therapy. HBeAg seroconversion may take place up to 6 months after discontinuation of interferon. HBeAg loss and seroconversion appear to be much more durable when induced with interferon in comparison with a nucleoside/nucleotide analogue. Interferon is most effective in patients with genotype A infection and least effective in those with genotypes D and C.
- If HBsAg titers are available, they can be used to guide interferon-based therapy. Discontinuation of interferon therapy is indicated in all patients with HBsAg > 20,000 IU/mL at week 24, irrespective of the HBV genotype [42]. Alternatively, those with no decline in the HBsAg titer at 12 weeks should also stop therapy. Stopping rules improve the cost-effectiveness of peginterferon therapy [43].
HBeAg-negative hepatitis
HBeAg-negative CHB represents a late phase in the course of chronic HBV infection.
- The patient should be considered for treatment if:
— HBV DNA ≥ 20,000 IU/mL and serum ALT > 2 × ULN
- Liver biopsy or other forms of fibrosis assessment should be considered in patients with:
— HBV DNA ≥ 20,000 IU/mL and serum ALT < 2 × ULN
— HBV DNA ≥ 2000 IU/mL and/or serum ALT > ULN
— Treatment should be administered if the liver biopsy shows moderate/severe necroinflammation or significant fibrosis (≥ F2)
- Treat any patient with cirrhosis who has detectable HBV DNA.
Recommendations for treatment:
- It is imperative to check for HIV coinfection before treatment, since all approved nucleoside/nucleotide analogues have activity against HIV and will rapidly lead to drug-resistant HIV if used as monotherapy.
- The treatment regimen can be conventional interferon, peginterferon alfa, or nucleoside/nucleotide analogues. Interferon-based therapy must not be used in the presence of liver failure.
- In patients with contraindications to interferon, such as decompensated cirrhosis or autoimmune disease, oral nucleoside/nucleotide analogues are recommended.
- The duration of interferon or peginterferon therapy is 1 year. If by week 12 HBsAg has not dropped, combined with a less than 2 log decline in HBV DNA, interferon therapy should be stopped, as a response is unlikely [42,44].
- For oral antiviral therapy, agents with a low resistance rate such as entecavir or tenofovir are preferred, particularly in patients with cirrhosis. However, where economic constraints are a consideration, therapy can be started with lamivudine (or telbivudine), with early adefovir add-on therapy or a switch to tenofovir when drug resistance is detected or when HBV DNA remains at ≥ 2000 IU/mL at week 24 of therapy.
- The optimal duration of antiviral therapy for HBeAg-negative CHB is not known, but long-term therapy is required—possibly lifelong, or until loss of HBsAg.
- Monitoring both biochemistry and HBV DNA every 3–6 months is recommended for assessing the treatment response and for early detection of drug resistance.
- A drug with a nonoverlapping resistance profile should be added (adefovir for lamivudine resistance) when drug resistance is detected.
- If ALT is elevated and HBV DNA levels are low (< 2000 IU/mL), other causes of inflammation (fatty liver, medication, coinfection with HDV and HCV) should be excluded. HDV inhibits HBV replication, and HDV-coinfected patients are therefore typically HBeAg-negative, with low or even undetectable levels of HBV DNA but persistently high ALT levels, often with evidence of advanced fibrosis/cirrhosis.
4.3 Coinfection
HBV–HDV
Hepatitis D virus (HDV) is a defective virus with a circular RNA genome and a single structured protein, the hepatitis delta antigen. The virus requires HBV surface antigen to serve as an envelope for its delta antigen. This helper function of HBV is required for HDV assembly and propagation.
- Up to 5% of the world’s population is infected with HBV, and probably 5% of those chronically infected with HBV have HDV infection.
- However, some endemic areas in the developing world may have much higher rates (Horn of Africa, Eastern Europe, Amazon Basin). The virus simultaneously coinfects with HBV, or superinfects in someone already chronically infected with HBV.
- Coinfection evolves to chronicity in only 2% of cases, but is associated with a higher chance of fulminant acute infection, while superinfection leads to progressive disease and cirrhosis in more than 80% of cases.
- Cirrhosis develops at a younger age than in patients with chronic HBV monoinfection.
Recommendations:
- Universal HBV vaccination should be implemented to prevent HDV infection in the community and thereby decrease its prevalence.
- HBsAg-positive patients should be evaluated to rule out HDV infection, particularly if hepatitis is present in the face of little or no HBV viral replication (i.e., a low HBV DNA), or if they come from an HDV-endemic region or have acquired HBV through injection drug use.
- HDV infection can be diagnosed by detection of HDV RNA in serum by polymerase chain reaction, or indirectly by detection of antibodies against hepatitis D antigen (anti-HDV) of the IgG and IgM classes.
- Chronic hepatitis D should be treated with interferon (preferably pegylated interferon) for at least 12 months, but the treatment results are suboptimal. Patients with active HBV replication despite HDV coinfection may benefit from treatment with nucleoside/nucleotide analogues (NAs) in combination with peginterferon.
HBV–HCV
Infection with HBV and hepatitis C (HCV) viruses may occur, as the two share similar risk factors and some common modes of transmission. Coinfection is most common in regions highly endemic for both viruses and in individuals who have contracted the infection through injection drug use—since unlike HBV, HCV is poorly transmitted via the sexual or vertical route. For the same reasons, HBV and HCV coinfection—and even triple infection with HBV, HCV and HIV and potentially quadruple infection (with HDV in addition)—may be observed in high-risk populations.
- The interferons (and pegylated interferons) are well-established therapeutic agents for both HBV and HCV and represent the treatment of choice for coinfected patients (in the absence of HIV).
- When HCV predominates (with detectable HCV RNA and low or undetectable HBV DNA), HCV therapy, which is rapidly evolving, should be prioritized. IFN-based therapy for HCV may be preferable in order to control HBV as well, but there are no robust data on this approach to date. New interferon-free therapies for HCV are highly effective and should be considered in HBV/HCV-coinfected patients. Optimal approaches for this population are being evaluated.
- When HBV predominates (with high HBV DNA levels), hepatitis C has often been cleared (i.e., undetectable HCV-RNA). In such cases, treatment decisions regarding HBV should be made irrespective of the presence of past HCV infection.
- Regular monitoring of ALT and of HCV RNA and HBV DNA during and after therapy is required, as suppression of the dominant virus by antiviral therapy may result in reactivation of the previously suppressed virus.
HBV–HIV
An estimated 36 million persons throughout the world are infected with HIV. Chronic coinfection with HBV may be present, due to the common modes of transmission of the viruses—parenteral, vertical, and sexual.
- The prevalence of CHB among HIV-infected persons may be ten times or more higher than that of the background population.
- Chronic HBV infection occurs in 5–10% of HIV-infected persons in Western Europe and the United States [45]
- Progression of CHB to cirrhosis, end-stage liver disease, and/or HCC is more rapid in HIV-infected persons than in persons with CHB alone [46].
The absence of controlled trials and the dual activity of some agents complicate the management of CHB infection in patients with HIV coinfection. Treatment regimens depend on the clinical status of both HIV and HBV.
- Many approved nucleoside/nucleotide analogues with activity against HBV also suppress HIV, and it is therefore critical that monotherapy with any approved oral HBV agents should be avoided, as resistance to HIV and possibly to HBV will rapidly occur. When treatment is indicated, a tenofovir-based regimen is preferred, in combination with other highly active agents for HIV.
- All patients with CHB should therefore always be checked for HIV coinfection before antiviral treatment is initiated.
The principal objectives of anti-HBV treatment are to stop or decrease the progression of liver disease, and to prevent cirrhosis and HCC.
- Prolonged suppression of HBV replication leads to histologic improvement, a significant decrease in or normalization of aminotransferases, and prevention of progression to cirrhosis and end-stage liver disease.
- Sustained viral control requires long-term maintenance therapy.
- Treatment discontinuation in particular may be associated with HBV reactivation and ALT flares.
- The drawback of long-term therapy is the risk of HBV resistance. To reduce drug resistance, most coinfected patients require HBV combination therapy.
4.4 Pregnancy
The following recommendations are also based on the 2012 EASL guideline [2]:
- All pregnant women should be screened for HBsAg.
- Before HBV treatment is started, the risk to the fetus in case of pregnancy and the patient’s family planning should be discussed.
- (PEG-)IFN is contraindicated during pregnancy.
- Tenofovir has a better resistance profile and more extensive safety data in pregnant, HBV-positive women than telbivudine (both are pregnancy category B drugs: no risk in animal studies, but unknown in humans) [47]. The data in HIV-positive pregnant women suggest that the use of lamivudine, emtricitabine, and tenofovir is safe [48,49].
- Perinatal HBV transmission mainly occurs at delivery, and prevention focuses on passive and active immunization with hepatitis B immunoglobulin (HBIg) and HBV vaccination, both of which must be given within 12 hours of birth.
- In a meta-analysis of the utility of HBIg given to newborns to prevent mother-to-child transmission (MTCT) of HBV, HBIg and HBV plasma–derived vaccine reduced transmission from 20% to 10% in comparison with plasma vaccine alone (RR 0.49; 95% CI, 0.32 to 0.74); with HBIg and recombinant HBV vaccine, transmission was reduced from 30.8% to 18.9% (RR 0.61; 95% CI, 0.41 to 0.92) [50].
- Women with high concentrations of HBV DNA (serum HBV DNA > 106–7 IU/mL, and mostly HBeAg-positive) may still have a high risk of MTCT despite appropriate vaccination and should be considered for treatment with lamivudine, telbivudine, or tenofovir during the last trimester of pregnancy, in addition to passive and active vaccination with HBIg and HBV vaccination.
- In a meta-analysis of RCTs, lamivudine reduced the transmission of HBV from 25.4% to 12% in comparison with a placebo when it was administered in late pregnancy. In comparison with patients who received HBIg, lamivudine reduced transmission from 20.4% to 6.3% [51]. In a meta-analysis of telbivudine treatment in pregnancy, the pooled results were similar to those with lamivudine, but the analysis only included two RCTs and three non-RCTs [52].
- NA therapy given only for the prevention of perinatal transmission may be discontinued within the first 3 months after delivery.
- HBV-infected women should be monitored closely after delivery, as flares may occur [53].
5. Hepatitis B vaccination
A program for universal vaccination of all newborns is a key step toward effective control of HBV infection throughout the world. HBV vaccination has been shown to be highly cost-effective. Vaccination prevents infection with HBV and thus reduces the incidence of chronic hepatitis, cirrhosis, and HCC in the vaccinated population, as well as reducing transmission by limiting the number of susceptible individuals.
5.1 Active vaccination with hepatitis B vaccine
HBsAg is the antigen used in the formulation of the hepatitis B vaccine. It is produced from yeast through recombinant DNA technology. It is available as a single-agent preparation or as a fixed combination with other vaccines.
5.2 Passive vaccination with hepatitis B immunoglobulin
Hepatitis B immunoglobulin (HBIg) is prepared from the plasma of individuals who have a high concentration of anti-HBs. The standard dose of HBIg is 0.06 mL/kg for all applications in adults or 200 IU in infants. In standard doses, it provides temporary protection (i.e., for approximately 3–6 months) against HBV infection. HBIg is administered by intramuscular injection, preferably into the deltoid or gluteal muscle. If it is given with hepatitis B vaccine, the HBIg vaccine should be administered at a different injection site.
5.3 Preexposure prophylaxis
A comprehensive strategy for eliminating HBV transmission should start with a preexposure vaccination program. This should include:
- Universal vaccination of all infants at birth; mandatory for infants born to pregnant women who test positive when screened for hepatitis B surface antigen.
- Postexposure immunoprophylaxis for children born to mothers whose HBsAg status is unknown.
- Catch-up vaccination of all children and adolescents who have not previously been vaccinated.
- Vaccination of unvaccinated adults exposed to risks of HBV infection (however, typically “high-risk” individuals frequently do not access health care or inform health-care facilities; hence the need for universal infant vaccination).
- Vaccination of those at risk of more severe infection—e.g., patients with chronic liver disease.
5.4 Vaccination schedules
The combination of prevalence, route of transmission, and viral factors has implications for the vaccination strategy—vaccination of at-risk groups, infant vaccination, or adolescent vaccination.
The vaccine is administered by intramuscular injection into the deltoid muscle (not the gluteal muscle) in adults, or into the anterolateral aspect of the thigh in neonates.
- Studies suggest that universal vaccination at birth is cost-effective in countries with high and moderate prevalence.
- Europe and North America, with very low incidence rates, have implemented either routine infant vaccination or vaccination for newborns of mothers who test positive for hepatitis B surface antigen (HBsAg).
- Routine adolescent vaccination at the age of 10 and catch-up vaccination for at-risk adults (it is difficult to identify and/or access those who are “at risk”) are recommended in some countries, but this will have little effect on the rate of chronic infection.
Vaccination recommendations:
- Primary vaccination, consisting of three or more intramuscular doses of hepatitis B vaccine administered at 0, 1, and 6 months, results in a positive antibody response in 30–55% of adults aged ≤ 40 years after the first dose, 75% after the second dose, and > 90% after the third dose. These response rates decline when the vaccine is given to older individuals (e.g., < 90% in persons > 40 years old, 75% in those over 60 years old).
- Other innovative vaccination schedules (e.g. 0, 1, and 4 months or 0, 2, and 4 months or 0, 1, and 2 months) are able to produce dose-specific and final rates of protection similar to those obtained with the 0, 1, 6-month schedule, and may be more practical for newborns.
- Accelerated vaccination schedules for postexposure prophylaxis in adults often ensure compliance with completion of the vaccination schedule.
- Babies born to HBsAg-positive mothers should receive the first dose of vaccine within 12 hours of birth.
- Host factors (e.g., smoking, obesity, cirrhosis, genetic factors, immune suppression, renal failure, etc.) are known to result in a decreased vaccine response.
- Individuals who do not mount an anti-HBs response (≥ 10 mIU/mL) to the primary vaccination schedule should receive a repeat three-dose vaccination (at 0, 1 and 2 months). This gives rise to protective antibody levels in 44–100% of individuals. Individuals who do not develop protective anti-HBs levels after revaccination can be considered for repeat vaccination (0, 1, and 2 months, with a 6-month booster) with double the standard dosage of vaccine.
- For persons ≥ 18 years old who do not live in an area endemic for hepatitis A, a combined hepatitis A–hepatitis B vaccine (Twinrix) is available.
5.5 Postexposure prophylaxis
Postexposure prophylaxis should be considered for individuals who have had recent exposure (either parenteral or sexual) to blood or other body fluids, if it can be carried out in a timely fashion.
- Evaluation of the HBsAg status of the infective source and the anti-HBs status of the exposed person should be carried out before the vaccine is administered.
- In countries with a high level of HBV endemicity, HBsAg in the exposed individual should also be checked.
- Individuals without prior vaccination should receive both HBIg and hepatitis B vaccine soon after exposure (preferably within 24 h). Hepatitis B vaccine administered simultaneously with HBIg must be at a different injection site.
- Completion of the hepatitis B vaccine series is again at 0, 1, and 6 months or 0, 1, and 2 months.
Exposed individuals who are in the process of being vaccinated (but who have not completed the vaccine series) should receive the appropriate dose of HBIg and should be advised to complete the hepatitis B vaccination series.
Vaccine responders may maintain protective anti-HBs levels for various lengths of time. Individuals who respond to hepatitis B vaccination are protected for at least 20 years (perhaps lifelong), even if vaccinees lack detectable anti-HBs at the time of a recent exposure. Asymptomatic acute hepatitis B infection can occur in vaccine responders following a decrease in anti-HBs levels, but it is usually self-limited. Occult hepatitis B infection has been recognized in some vaccinated patients, but the clinical significance of this is unclear [54].
Thus, immunocompetent persons who are known to have responded to hepatitis B vaccination with anti-HBs concentrations of ≥ 10 mIU/mL do not require additional passive or active immunization after an HBV exposure. In addition, they do not need further periodic testing to assess anti-HBs concentrations. However, if the previous anti-HBs concentration is not known (not routinely tested) or is < 10 mIU/mL, then HBIg and hepatitis B vaccine should be given. If the exposed individual is a known nonresponder, then two doses of HBIg, 1 month apart, can be given.
Booster doses are not recommended routinely for immunocompetent individuals, whether they have received the vaccination as infants, adolescents, or adults. Likewise, serologic testing to assess antibody concentrations in any age group is not recommended, except perhaps for individuals at high risk of infection such as household contacts of infected persons or health-care workers—e.g., a booster dose should be administered when the anti-HBs level is < 10 mIU/mL. It is prudent to recommend booster doses to individuals with a clear, ongoing risk of HBV infection (e.g., when the sexual partner is HBsAg-positive, or in health-care personnel).
5.6 Pregnancy and hepatitis B vaccination
There are no teratogenic or other risks to the fetus if hepatitis B vaccine is administered to pregnant women. There are no contraindications for hepatitis B vaccination or HBIg administration in pregnant or lactating mothers.
6. Appendix
6.1 Abbreviations
| AASLD |
American Association for the Study of Liver Diseases |
| AFP |
Alpha fetoprotein |
| ALT |
Alanine aminotransferase |
| AST |
Aspartate aminotransferase |
| APASL |
Asian–Pacific Association for the Study of the Liver |
| BCP |
Basal core promoter |
| CBC |
Complete blood count |
| CHB |
Chronic hepatitis B |
| CI |
Confidence interval(s) |
| EASL |
European Association for the Study of the Liver |
| FDA |
Food and Drug Administration (United States) |
| HBc |
Hepatitis B core (antigen) |
| HBeAg |
Hepatitis B extracellular antigen |
| HBIg |
Hepatitis B immunoglobulin |
| HBsAg |
Hepatitis B surface antigen |
| HBV |
Hepatitis B virus |
| HCC |
Hepatocellular carcinoma |
| HCV |
Hepatitis C virus |
| HDV |
Hepatitis D virus |
| HIV |
Human immunodeficiency virus |
| IFN |
Interferon |
| IgG |
Immunoglobulin G |
| IgM |
Immunoglobulin M |
| INR |
International normalized ratio |
| IU/mL |
International units per milliliter (the WHO standard for HBV DNA concentrations) |
| LLD |
Lower limit of detection |
| MTCT |
Mother-to-child transmission |
| NA |
Nucleoside analogue |
| NAFLD |
Nonalcoholic fatty liver disease |
| NASH |
Nonalcoholic steatohepatitis |
| NICE |
National Institute for Care and Health Excellence |
| NSAID |
Nonsteroidal anti-inflammatory drug |
| PCR |
Polymerase chain reaction |
| PEG-IFN |
Peginterferon |
| RCT |
Randomized controlled trial |
| RR |
Relative risk |
| SCS |
Systemic corticosteroids |
| ULN |
Upper limit of normal |
| WGO |
World Gastroenterology Organisation |
| WHO |
World Health Organization |
For a definition of frequently used terms, reference may be made to page 533 of the 2012 APASL guideline [4].
6.2 References
- Lok ASF, McMahon BJ. Chronic hepatitis B: update 2009. Hepatology 2009;50:661–2.
- European Association for the Study of the Liver. EASL clinical practice guidelines: management of chronic hepatitis B virus infection. J Hepatol 2012;57:167–85.
- National Institute for Health and Care Excellence (NICE). Hepatitis B (chronic): diagnosis and management of chronic hepatitis B in children, young people and adults [Internet]. London: NICE; 2013. Available from: http://www.nice.org.uk/guidance/cg165/resources/guidance-hepatitis-b-chronic-pdf
- Liaw YF, Kao JH, Piratvisuth T, Chan HLY, Chien RN, Liu CJ, et al. Asian–Pacific consensus statement on the management of chronic hepatitis B: a 2012 update. Hepatol Int 2012;6:531–61.
- World Health Organization. Global Alert and Response (GAR): Hepatitis [Internet]. Geneva: World Health Organization [cited 2015 Mar 31]. Available from: http://www.who.int/csr/disease/hepatitis/en/.
- Ott JJ, Stevens GA, Groeger J, Wiersma ST. Global epidemiology of hepatitis B virus infection: new estimates of age-specific HBsAg seroprevalence and endemicity. Vaccine 2012;30:2212–9.
- Hepatitis B Foundation. Hepatitis B Foundation [Internet]. Doylestown, PA. Available from: http://www.hepb.org/.
- Lozano R, Naghavi M, Foreman K, Lim S, Shibuya K, Aboyans V, et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012;380:2095–128.
- Hollinger F, Liang T. Hepatitis B virus. In: Knipe DM, Howley PM, editors. Fields’ virology. 4th ed. Philadelphia: Lippincott Williams & Wilkins; 2001. p. 2971–3036.
- Sunbul M. Hepatitis B virus genotypes: global distribution and clinical importance. World J Gastroenterol 2014;20:5427.
- Shi W, Zhang Z, Ling C, Zheng W, Zhu C, Carr MJ, et al. Hepatitis B virus subgenotyping: history, effects of recombination, misclassifications, and corrections. Infect Genet Evol 2013;16:355–61.
- Chulanov V, Neverov A, Karandashova I, Dolgin V, Mikhailovskaya G, Lebedeva E, et al. Molecular epidemiology of HBV in Russia [abstract C.222]. Abstracts of the 14th International Symposium on Viral Hepatitis and Liver Disease. China, Shanghai, 2012.
- Deterding K, Constantinescu I, Nedelcu FD, Gervain J, Nemecek V, Srtunecky O, et al. Prevalence of HBV genotypes in Central and Eastern Europe. J Med Virol 2008;80:1707–11.
- Devesa M, Loureiro CL, Rivas Y, Monsalve F, Cardona N, Duarte MC, et al. Subgenotype diversity of hepatitis B virus American genotype F in Amerindians from Venezuela and the general population of Colombia. J Med Virol 2008;80:20–6.
- Blitz L, Pujol FH, Swenson PD, Porto L, Atencio R, Araujo M, et al. Antigenic diversity of hepatitis B virus strains of genotype F in Amerindians and other population groups from Venezuela. J Clin Microbiol 1998;36:648–51.
- Cardona NE, Loureiro CL, Garzaro DJ, Duarte MC, García DM, Pacheco MC, et al. Unusual presentation of hepatitis B serological markers in an Amerindian community of Venezuela with a majority of occult cases. Virol J 2011;8:527.
- Pujol FH, Navas MC, Hainaut P, Chemin I. Worldwide genetic diversity of HBV genotypes and risk of hepatocellular carcinoma. Cancer Lett 2009;286:80–8.
- Kramvis A, Kew M, François G. Hepatitis B virus genotypes. Vaccine 2005;23:2409–23.
- Kimbi GC, Kramvis A, Kew MC. Distinctive sequence characteristics of subgenotype A1 isolates of hepatitis B virus from South Africa. J Gen Virol 2004;85:1211–20.
- Kramvis A, Kew MC. Epidemiology of hepatitis B virus in Africa, its genotypes and clinical associations of genotypes. Hepatol Res 2007;37(s1):S9–S19.
- Yousif M, Mudawi H, Bakhiet S, Glebe D, Kramvis A. Molecular characterization of hepatitis B virus in liver disease patients and asymptomatic carriers of the virus in Sudan. BMC Infect Dis 2013;13:328.
- Devesa M, Pujol FH. Hepatitis B virus genetic diversity in Latin America. Virus Res 2007;127:177–84.
- Ahmed CS, Wang Z, Bin Z, Chen J, Kamal M, Hou J. Hepatitis B virus genotypes, subgenotypes, precore, and basal core promoter mutations in the two largest provinces of Pakistan. J Gastroenterol Hepatol 2009;24:569–73.
- De Franchis R, Hadengue A, Lau G, Lavanchy D, Lok A, McIntyre N, et al. EASL International Consensus Conference on Hepatitis B. 13–14 September, 2002 Geneva, Switzerland. Consensus statement (long version). J Hepatol 2003;39 Suppl 1:S3–25.
- McMahon BJ. Epidemiology and natural history of hepatitis B. Semin Liver Dis 2005;25 Suppl 1:3–8.
- World Health Organization. Hepatitis B. WHO fact sheet no. 204 [Internet]. Geneva: World Health Organization; 2015. Available from: http://www.who.int/mediacentre/factsheets/fs204/en/.
- Santantonio T, Fasano M. Current concepts on management of chronic hepatitis B. In: Serviddio G, editor. Practical management of chronic viral hepatitis [Internet]. InTech; 2013 [cited 2015 Mar 31]. Available from: http://www.intechopen.com/books/practical-management-of-chronic-viral-hepatitis/current-concepts-on-management-of-chronic-hepatitis-b.
- Hui CK, Leung N, Yuen ST, Zhang HY, Leung KW, Lu L, et al. Natural history and disease progression in Chinese chronic hepatitis B patients in immune-tolerant phase. Hepatology 2007;46:395–401.
- Brunetto MR, Oliveri F, Colombatto P, Moriconi F, Ciccorossi P, Coco B, et al. Hepatitis B surface antigen serum levels help to distinguish active from inactive hepatitis B virus genotype D carriers. Gastroenterology 2010;139:483–90.
- Raimondo G, Allain JP, Brunetto MR, Buendia MA, Chen DS, Colombo M, et al. Statements from the Taormina expert meeting on occult hepatitis B virus infection. J Hepatol 2008;49:652–7.
- Zerbini A, Pilli M, Boni C, Fisicaro P, Penna A, Di Vincenzo P, et al. The characteristics of the cell-mediated immune response identify different profiles of occult hepatitis B virus infection. Gastroenterology 2008;134:1470–81.
- Peng CY, Chien RN, Liaw YF. Hepatitis B virus-related decompensated liver cirrhosis: benefits of antiviral therapy. J Hepatol 2012;57:442–50.
- Fattovich G, Bortolotti F, Donato F. Natural history of chronic hepatitis B: special emphasis on disease progression and prognostic factors. J Hepatol 2008;48:335–52.
- Lee IC, Lin CH, Huang YH, Huo TI, Su CW, Hou MC, et al. IL28B polymorphism correlates with active hepatitis in patients with HBeAg-negative chronic hepatitis B. PloS One 2013;8:e58071.
- Kim TW, Kim MN, Kwon JW, Kim KM, Kim SH, Kim W, et al. Risk of hepatitis B virus reactivation in patients with asthma or chronic obstructive pulmonary disease treated with corticosteroids. Respirology 2010;15:1092–7.
- Huang YH, Hsiao LT, Hong YC, Chiou TJ, Yu YB, Gau JP, et al. Randomized controlled trial of entecavir prophylaxis for rituximab-associated hepatitis B virus reactivation in patients with lymphoma and resolved hepatitis B. J Clin Oncol 2013;31:2765–72.
- Singal AG, Conjeevaram HS, Volk ML, Fu S, Fontana RJ, Askari F, et al. Effectiveness of hepatocellular carcinoma surveillance in patients with cirrhosis. Cancer Epidemiol Biomark Prev 2012;21:793–9.
- Liaw YF, Leung N, Kao JH, Piratvisuth T, Gane E, Han KH, et al. Asian–Pacific consensus statement on the management of chronic hepatitis B: a 2008 update. Hepatol Int 2008;2:263–83.
- Lok ASF, McMahon BJ. Chronic hepatitis B. Hepatology 2007;45:507–39.
- Hepatitis B Foundation. HBF drug watch: compounds in development for chronic hepatitis B. Updated December 15, 2014 [Internet]. Available from: http://www.hepb.org/professionals/hbf_drug_watch.htm
- World Health Organization. Global policy report on the prevention and control of viral hepatitis in WHO member states [Internet]. Geneva: World Health Organization; 2013. Available from: http://www.who.int/hiv/pub/hepatitis/global_report/en/
- Sonneveld MJ, Hansen BE, Piratvisuth T, Jia JD, Zeuzem S, Gane E, et al. Response-guided peginterferon therapy in hepatitis B e antigen-positive chronic hepatitis B using serum hepatitis B surface antigen levels. Hepatology 2013;58:872–80.
- Lo AOS, Wong VWS, Wong GLH, Chan HLY, Dan YY. Cost effectiveness of response-guided therapy with peginterferon in the treatment of chronic hepatitis B. Clin Gastroenterol Hepatol 2015;13:377–85.
- Sonneveld MJ, Rijckborst V, Boucher CAB, Hansen BE, Janssen HLA. Prediction of sustained response to peginterferon alfa-2b for hepatitis B e antigen-positive chronic hepatitis B using on-treatment hepatitis B surface antigen decline. Hepatology 2010;52:1251–7.
- Spradling PR, Richardson JT, Buchacz K, Moorman AC, Brooks JT, HIV Outpatient Study (HOPS) Investigators. Prevalence of chronic hepatitis B virus infection among patients in the HIV Outpatient Study, 1996–2007. J Viral Hepat 2010;17:879–86.
- Thio CL, Seaberg EC, Skolasky R, Phair J, Visscher B, Muñoz A, et al. HIV-1, hepatitis B virus, and risk of liver-related mortality in the Multicenter Cohort Study (MACS). Lancet 2002;360:1921–6.
- Borgia G, Carleo MA, Gaeta GB, Gentile I. Hepatitis B in pregnancy. World J Gastroenterol 2012;18:4677–83.
- Terrault NA, Jacobson IM. Treating chronic hepatitis B infection in patients who are pregnant or are undergoing immunosuppressive chemotherapy. Semin Liver Dis 2007;27 Suppl 1:18–24.
- Chotiyaputta W, Lok AS. Role of antiviral therapy in the prevention of perinatal transmission of hepatitis B virus infection. J Viral Hepat 2009;16:91–3.
- Lee C, Gong Y, Brok J, Boxall EH, Gluud C. Effect of hepatitis B immunisation in newborn infants of mothers positive for hepatitis B surface antigen: systematic review and meta-analysis. BMJ 2006;332:328–36.
- Han L, Zhang HW, Xie JX, Zhang Q, Wang HY, Cao GW. A meta-analysis of lamivudine for interruption of mother-to-child transmission of hepatitis B virus. World J Gastroenterol 2011;17:4321–33.
- Deng M, Zhou X, Gao S, Yang SG, Wang B, Chen HZ, et al. The effects of telbivudine in late pregnancy to prevent intrauterine transmission of the hepatitis B virus: a systematic review and meta-analysis. Virol J 2012;9:185.
- Ter Borg MJ, Leemans WF, de Man RA, Janssen HLA. Exacerbation of chronic hepatitis B infection after delivery. J Viral Hepat 2008;15:37–41.
- Lai MW, Lin TY, Tsao KC, Huang CG, Hsiao MJ, Liang KH, et al. Increased seroprevalence of HBV DNA with mutations in the s gene among individuals greater than 18 years old after complete vaccination. Gastroenterology 2012;143:400–7.

