Gut Microbes
 |
Virginia Robles-Alonso, MD
Digestive System Research Unit
University Hospital Vall d’Hebron, Ciberehd
Barcelona, Spain
|
| |
|
 |
Francisco Guarner, MD
Digestive System Research Unit
University Hospital Vall d’Hebron, Ciberehd
Barcelona, Spain
|
Microbial communities: different approach, novel insights
Until recent years, our knowledge on the human gut microbiota was largely limited to certain community members with potential pathogenicity by either translocation or production of toxins. Most of these potential pathogens were isolated in culture and recognized by traditional diagnostic techniques. However, culture-based techniques to identify bacteria have important limitations, and the large majority of bacteria in the human gut are considered ‘unculturable’ for the great difficulties for growing them in the laboratory. Their potential role in health or disease has been ignored.
The notion of a host-microbe symbiotic relationship in the gut, in terms of proven benefits or mutualism among partners, is supported by studies carried out with animals born and bred under germ-free conditions 1. Compared with colonized counterparts, germ-free mammalians or birds exhibit major differences in body anatomy and physiology. These studies clearly indicated that microbial communities colonizing the animal host have a strong influence on body growth and development, as well as on the induction and regulation of the immune system, thus contributing to maintenance of health during life. However, very little is known about nature and biological characteristics of the critical symbionts inducing beneficial effects on the animal host.
The advent of high-throughput technologies has changed our perspective dramatically. First, these technologies are culture independent and, remarkably, they allow the characterization of microbial communities as a whole, enabling a deeper and global view of all the community members and their relative abundance 2. The novel approach for the analysis of microbial communities in environmental samples is called “metagenomics”, and is defined as the study of all the genetic material recovered directly from environmental samples bypassing the need to isolate and culture individual community members 3. The metagenome is the collective genetic content of the combined genomes of the constituents of an ecological community. The microbiome is defined as the collective genome of the microbial symbionts in a host animal 4.
The most common approach consists of the extraction of DNA from the biological sample, followed by the amplification and sequencing of 16S ribosomal RNA genes in the sample. The 16S rRNA gene is present in all bacteria and contains both conserved and variable regions. Thus, similarities and differences in the sequence of nucleotides of the 16S rRNA gene allow taxonomic identification ranging from the domain and phylum level to the species level. Currently, around three million aligned and annotated 16S rRNA sequences are available in DNA databases (http://rdp.cme.msu.edu/). Taxonomic identification is based on comparison of 16S rRNA sequences in the sample with reference sequences in the database. In this way, studies on the 16S rRNA gene provide information about microbial composition and diversity of species in a given sample.
The most powerful molecular approach is not limited to 16S rRNA sequencing but addresses all the genetic material in the sample. The decreasing cost and increasing speed of DNA sequencing, coupled with the advances in computational analyses of large datasets, have made it feasible to analyze complex mixtures of entire genomes with reasonable coverage. The resulting information describes the collective genetic content of the community from which functional and metabolic networks can be inferred. Thus, the full metagenomic approach has the advantage of not only providing the phylogenetical characterization of the microbial community but also telling about biological functions present in the community.
The Gut Microbiota
Estimates suggest that the colon, the largest ecological niche for microbial communities in the human body, harbors over 1014 microbial cells, i.e. several hundred grams of microbes, most of them belonging to the domain Bacteria. Molecular studies based on 16S rRNA gene sequencing have highlighted that only seven to nine of the 55 known divisions or phyla of the domain Bacteria are detected in fecal or mucosal samples from the human gut 4-7. Moreover, such studies also revealed that more than 90% of all the phylotypes belong to just two divisions: Bacteroidetes and Firmicutes (Figure 1). The other divisions that have been consistently found in samples from the human distal gut are Proteobacteria, Actinobacteria, Fusobacteria, and Verrucomicrobia. Of the 13 divisions of the domain Archea, only one or two species seem to be represented in the human distal gut microbiota. Thus, at the division level, the human intestinal ecosystem is less diverse than other ecosystems on earth, like soils and ocean waters which may contain 20 or more divisions 5. However, at a lower taxonomic level (species or strain), there is a considerable variation in the composition of the fecal microbiota among human individuals. Strain diversity between individuals is highly remarkable so that studies have found that a large proportion of the identified strain-level phylotypes are unique to each person 5. Each individual harbors his or her own distinctive pattern of bacterial composition.
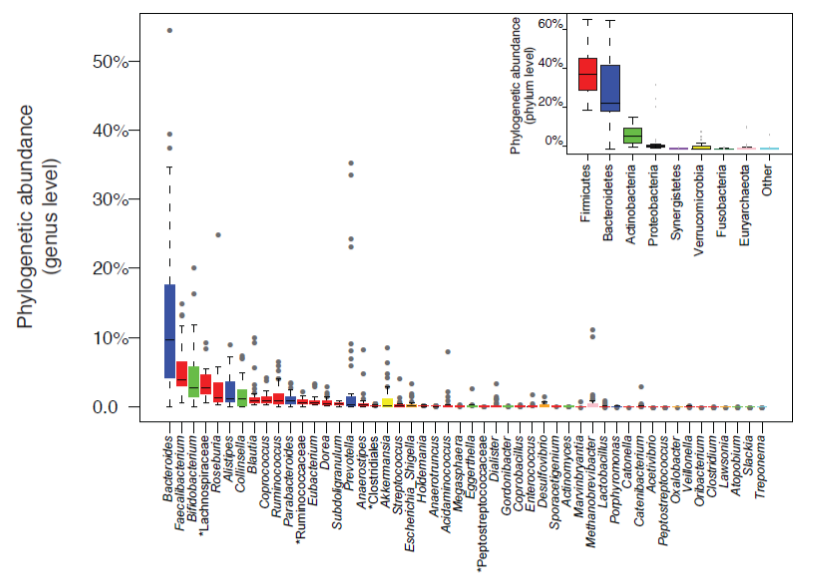
Figure 1: Genus abundance variation box plot for the 30 most abundant genera of the human gut microbiota as determined by metagenomic sequencing of human fecal samples. Genera are colored by their respective phylum (see inset for color key). Inset shows phylum abundance box plot. Genus and phylum level abundances were measured using reference-genome-based-mapping (Source: from Figure 1b in: Arumugam M et al, Enterotypes of the human gut microbiome. Nature 2011; 473:174-180; with permission).
In a cohort of 124 European adult subjects, a total of 3.3 million microbial genes were characterized by full metagenomic analysis of fecal samples 6. This effort has provided the first gene catalogue of the human gut microbiome. Each individual carries an average of 600,000 non-redundant microbial genes in the gastrointestinal tract. This figure suggests that most of the 3.3 million genes in the catalogue are shared. It was found that around 300,000 microbial genes are common in the sense that they are present in at least 50% of individuals. Up to 98% of genes in the catalogue are bacterial, and the entire cohort of individuals harbors between 1,000 and 1,150 prevalent bacterial species, with at least 160 species per individual6. Interestingly, Bacteroides, Faecalibacterium and Bifidobacterium are the most abundant genera but their relative proportion is highly variable across individuals (Figure 1).
Network analysis of species abundance across different individuals suggested that the overall structure of the human gut microbiota in each individual conforms to discrete and distinct patterns defined by interactions within community members. This hypothesis was investigated using a dataset of metagenomic sequences from American, European and Japanese individuals. The phylogenetic analysis for taxonomic assignments was performed by mapping the metagenomic sequences to the reference genomes of fully sequenced bacteria. Multidimensional cluster analysis and principal component analysis revealed that all individual samples formed three robust clusters, which have been designated as ‘enterotypes’ 7. Each of the three enterotypes is identifiable by the variation in the levels of one of three genera: Bacteroides (enterotype 1), Prevotella (enterotype 2) and Ruminococcus (enterotype 3). The basis for the enterotype clustering is unknown but appears independent of nationality, sex, age, or body mass index. The enterotype concept suggests that enteric microbiota variations across individuals are generally stratified, not continuous. This further indicates the existence of a limited number of well-balanced host-microbial symbiotic states.
Interestingly, it seems that the reported enterotype partitioning is related to long-term dietary patterns 8. The Bacteroides enterotype was associated with diets enriched in protein and fat. In contrast, the Prevotella enterotype was linked to diets with predominance of carbohydrates and sugars.
Sequencing analysis has allowed describing not only differences in microbial communities between humans but also intra-individual variability (Figure 2). Factors such as diet, drug intake or traveling may have an impact on microbial composition over time in a unique host. A recent study 9 collected samples from three different body sites (gut, mouth and skin) of two healthy subjects on a daily basis for a period of 15 and six months, respectively. Community differentiation by body site is highly stable over time but, within the same body site, a low stability across time was noted. At species level, very few microbial members would constitute the so-called ‘core human gut microbiota’ 10, since only 5% of species were always present in all samples from the same individual.
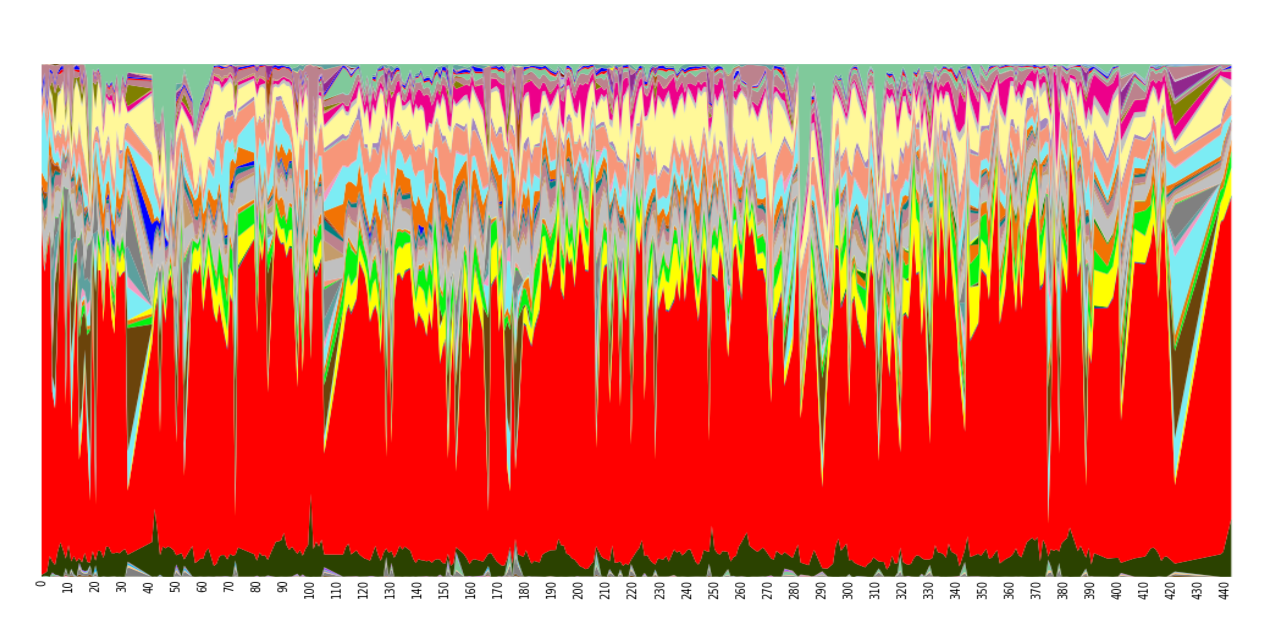
Figure 2: Temporal variation in genus abundance in fecal samples from a single human individual, who was sampled daily for 15 months. Columns represent microbial composition of each sample at genus level, and colors indicate genera as follows: Bacteroides, red; Faecalibacterium, beige; Akkermansia, pale green; Roseburia, light blue; Parabacteroides, yellow; other bacteroides, black; Bifidobacterium, grey, etc. (Source: from Additional file 8 in: Caporaso JG, Lauber CL, Costello EK et al. (2011) Genome Biol 12(5):R50; with permission).
Dysbiosis as a cause of disease
An imbalance of the normal gut microbiota composition is called dysbiosis. Different diseases have been associated with changes in the composition of the gut microbiota.
The world is facing a global health crisis provoked by an obesity epidemic. The incidence of malignant forms of obesity that are associated with cardiovascular complications, the metabolic syndrome, is steadily increasing in Western countries. Some recent data on the metabolic syndrome suggest that changes in gut microbiome composition may play a role in the disorder. Studies performed in mice revealed a shift in the abundance of Bacteroidetes and Firmicutes 11. In addition, recent human studies have shown that low genetic diversity in the gut microbiome increases the risk of several features associated with the metabolic disorders 12. The study recruited obese and non-obese individuals and those with a low bacterial gene count showed an inflammatory profile (increased C-reactive protein), disturbed glucose homeostasis (hyperglycemia, insulin resistance) as well as body fat accumulation (leptin resistance). Surprisingly, those alterations were linked to reduced microbial diversity independently of the presence of obesity or not. A subsequent study 13 revealed that a low-caloric diet including prebiotics (5-6g of inulin per day) increases the diversity of the intestinal microbiota, and reduces inflammatory abnormalities.
In animal models, transplantation of gut microbiota from obese mice to non-obese, germ-free mice resulted in transfer of metabolic syndrome-associated features from the donor to the recipient 14. The mechanisms advocated are the provision of additional energy by the conversion of dietary fiber to short-chain fatty acids, effects on gut-hormone production, and increased intestinal permeability causing elevated systemic levels of lipopolysaccharides. The contact with these antigens seems to contribute to low-grade inflammation, a characteristic trait of obesity and the metabolic syndrome. Presumably, obesity affects the diversity of the gut microbiota and probably, the way individuals harvest energy from nutrients.
The possible role of the intestinal metabolome on the development of cardiovascular risk is another hot issue. Phosphatidylcholine is a phospholipid involved in several metabolic functions, including the synthesis of neurotransmitters and amino acids, or structural features, such as being part of the cell membrane. Recent studies show that some members of the gut microbiota can degrade phosphatidylcholine to toxic metabolites, in particular to trimethylamine N-oxide (TMAO) 15, whose plasma levels are directly related to risk of cardiovascular events. In human studies, administration of a course of broad spectrum antibiotics decreases TMAO levels, demonstrating the contribution of some gut microbes to plasma TMAO. These findings reinforce the concept the microbiota is a metabolic organ that in addition to beneficial effects may also be involved in certain pathophysiological mechanisms.
One of the major hypotheses underlying the pathogenesis of inflammatory bowel disease (IBD) is the presence of abnormal communication between gut microbial communities and the mucosal immune system 16. Luminal bacteria appear to provide the stimulus for immune-inflammatory responses leading to mucosal injury. There is also some evidence showing that the microbiota of patients with IBD differs from that of healthy subjects. Differences include low biodiversity of dominant bacteria, high variability over time, and changes both in composition and spatial distribution (high concentrations of mucosa-adherent bacteria). The microbiota of Crohn’s disease patients is characterized by a decrease in Faecalibacterium prausnitzii 17 as well as increased numbers of the Proteobacteria and Actinobacteria phyla 18. Some other associations of human conditions with particular microbiota characteristics have been described such as irritable bowel syndrome, psoriasis, colorectal carcinoma and childhood-onset asthma, but consistency among studies is still poor.
Irritable bowel syndrome (IBS) is another chronic digestive disorder. Although no structural abnormalities have been demonstrated, molecular changes such as increased permeability of the intestinal barrier and increased visceral sensitivity are common features in IBS sufferers. Some evidence suggests that gut microbes can be involved in the origin of such alterations. Recent data suggest decreased diversity in small-bowel microbiota of patients with IBS, with increased abundance of gram-negative organisms 19.
Microbial therapeutics
Even if associations of dysbiosis with disease do not necessarily predict cause-effect relationships, there is growing interest to develop strategies that will improve the ‘physiological’ quality of the human gut microbial ecosystem for health benefits. As suggested by experts, the future of a healthy human gut microbiota may include the restoration of our ancestral microbial ecology. According to Cho and Blaser 20 there are two possible types of restoration. The first involves restoring ancient organisms in healthy hosts that lack them, as prophylaxis against future risk of disease. The second type of restoration could be therapeutic, when the etiological extinctions or imbalances are clearly identified. This scientific boundary will require an understanding of the biology of re-introductions, as well as developing microbial breeding programs 20.
Different interventional approaches have emerged, including the use of antibiotics, probiotics, prebiotics, combinations of probiotics and prebiotics, or techniques for microbial reconstitution by fecal transplantation. The referred approaches aim at improving host-microbes symbiosis in the gut by combating overgrowth of opportunistic community members or providing live microorganism or metabolic substrates in order to promote growth and activity of beneficial species.
Probiotics were defined as “live micro-organisms which, when administered in adequate amounts as part of food, confer a health benefit on the host” as proposed by the Joint FAO/WHO Expert Consultation in 2001. Our guideline for the use of probiotics and prebiotics in gastroenterology is available online 21, and will be updated later this year.
The term prebiotic refers to “a selectively fermented ingredient that allows specific changes, both in the composition and/or activity in the gastrointestinal microbiota that confers benefits upon host well being and health”. Concurrently, a prebiotic should not be hydrolyzed by human intestinal enzymes, it should be selectively fermented by beneficial bacteria, and this selective fermentation should result in beneficial effects on health or well-being of the host 22.
Finally, fecal transplant has emerged as an alternative approach to treat relapsing diarrhea by Clostridium difficile infection. This procedure has shown success in a subset of patients who failed standard treatment, with reported response rates up to 87% 23. A total of 239 patients who had undergone fecal transplantation were reported. Seventeen of 22 studies of fecal transplantation were performed in patients with fulminant or refractory Clostridium difficile infection. The major concern about this approach is the potential risk of transmitting infectious diseases 23.
Conclusions
The development of novel gene sequencing technologies as well as the availability of powerful bio-informatic analysis tools have allowed a dramatic proliferation of research work on the human gut microbiota. Large-scale studies are providing a deeper insight on the microbial communities that usually inhabit the human gut, and allow the identification of changes that are associated with disease states. A better knowledge of the contributions of microbial symbionts to host health will certainly help in the design of new potential interventions to improve symbiosis and combat disease. Moreover, such sequencing techniques provide novel insights into the field of infectious diseases by enabling the discovery of microbial pathogens, more accurate diagnostic tests, and disclosure of drug-resistance profiles 24.
References
- Wostmann BS. The germfree animal in nutritional studies. Annual review of nutrition. 1981;1:257-79. Epub 1981/01/01.
- Handelsman J, Rondon MR, Brady SF, Clardy J, Goodman RM. Molecular biological access to the chemistry of unknown soil microbes: a new frontier for natural products. Chemistry & biology. 1998;5(10):R245-9. Epub 1998/11/18.
- Frank DN, Pace NR. Gastrointestinal microbiology enters the metagenomics era. Current opinion in gastroenterology. 2008;24(1):4-10. Epub 2007/11/29.
- Turnbaugh PJ, Ley RE, Hamady M, Fraser-Liggett CM, Knight R, Gordon JI. The human microbiome project. Nature. 2007;449(7164):804-10. Epub 2007/10/19.
- Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, et al. Diversity of the human intestinal microbial flora. Science. 2005;308(5728):1635-8. Epub 2005/04/16.
- Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464(7285):59-65. Epub 2010/03/06.
- Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, Mende DR, et al. Enterotypes of the human gut microbiome. Nature. 2011;473(7346):174-80. Epub 2011/04/22.
- Wu GD, Chen J, Hoffmann C, Bittinger K, Chen YY, Keilbaugh SA, et al. Linking longterm dietary patterns with gut microbial enterotypes. Science. 2011;334(6052):105-8. Epub 2011/09/03.
- Caporaso JG, Lauber CL, Costello EK, Berg-Lyons D, Gonzalez A, Stombaugh J, et al. Moving pictures of the human microbiome. Genome biology. 2011;12(5):R50. Epub 2011/06/01.
- Costello EK, Lauber CL, Hamady M, Fierer N, Gordon JI, Knight R. Bacterial community variation in human body habitats across space and time. Science. 2009;326(5960):1694-7. Epub 2009/11/07.
- Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature. 2006;444(7122):1022-3. Epub 2006/12/22.
- Le Chatelier E, Nielsen T, Qin J, Prifti E, Hildebrand F, Falony G, et al. Richness of human gut microbiome correlates with metabolic markers. Nature. 2013;500(7464):541-6. Epub 2013/08/30.
- Cotillard A, Kennedy SP, Kong LC, Prifti E, Pons N, Le Chatelier E, et al. Dietary intervention impact on gut microbial gene richness. Nature. 2013;500(7464):585-8. Epub 2013/08/30.
- Blaut M, Klaus S. Intestinal microbiota and obesity. Handbook of experimental pharmacology. 2012(209):251-73. Epub 2012/01/18.
- Tang WH, Wang Z, Levison BS, Koeth RA, Britt EB, Fu X, et al. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. The New England journal of medicine. 2013;368(17):1575-84. Epub 2013/04/26.
- Guarner F. What is the role of the enteric commensal flora in IBD? Inflammatory bowel diseases. 2008;14 Suppl 2:S83-4. Epub 2008/09/26.
- Sokol H, Pigneur B, Watterlot L, Lakhdari O, Bermudez- Humaran LG, Gratadoux JJ, et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(43):16731-6. Epub 2008/10/22.
- Frank DN, St Amand AL, Feldman RA, Boedeker EC, Harpaz N, Pace NR. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(34):13780-5. Epub 2007/08/19.
- Mayer EA, Savidge T, Shulman RJ. Brain-gut microbiome interactions and functional bowel disorders. Gastroenterology. 2014;146(6):1500-12. Epub 2014/03/04.
- Cho I, Blaser MJ. The human microbiome: at the interface of health and disease. Nature reviews Genetics. 2012;13(4):260-70. Epub 2012/03/14.
- WGO Practice Guideline - Probiotics and Prebiotics. http://www.worldgastroenterology.org/probiotics-prebiotics.html.
- Coppa GV, Bruni S, Morelli L, Soldi S, Gabrielli O. The first prebiotics in humans: human milk oligosaccharides. Journal of clinical gastroenterology. 2004;38(6 Suppl):S80-3. Epub 2004/06/29.
- Landy J, Al-Hassi HO, McLaughlin SD, Walker AW, Ciclitira PJ, Nicholls RJ, et al. Review article: faecal transplantation therapy for gastrointestinal disease. Alimentary pharmacology & therapeutics. 2011;34(4):409-15. Epub 2011/06/21.
- Relman DA. Microbial genomics and infectious diseases. The New England journal of medicine. 2011;365(4):347-57. Epub 2011/07/29.
Glossary
Dysbiosis: An imbalance of the normal gut microbiota composition.
Enterotype: A classification of the human gut microbial communities into three groups or types, on the basis of the bacteriological composition of the ecosystem (diversity and abundance of the predominant genera).
Metagenome: The total genetic content of the combined genomes of the constituents of an ecological community.
Metagenomics: The study of all the genetic material recovered directly from environmental samples bypassing the need to isolate and
culture individual community members.
Microbiome: The collective genome of the microbial symbionts in a host animal.
Microbiota: The collection of microbial communities colonizing a particular ecological niche.
Phylotype: A microbial group defined by 16S rRNA sequence similarity rather than by phenotypic characteristics. A similarity of 97%
indicates approximately a specieslevel.
Symbionts: The microbial partners in symbiosis.
Symbiosis: Close and persistent interactions between living organisms of different species. Biological interactions may be mutualistic (both partners derive a benefit), commensalistic (one partner benefits without affecting the other), or parasitic (one benefits while the other is harmed). Most scientists believe that the term symbiosis should only refer to mutualistic relationships.